


北京基因組所(國家生物信息中心)合作發現罕見遺傳性血液病HLH的新致病基因
噬血細胞綜合征(HLH)是一種嚴重威脅生命的罕見血液病,發病率僅百萬分之一,因患者骨髓中常見吞噬血細胞現象而命名。HLH本質是由先天或后天因素導致的免疫系統過度激活,分泌過量細胞因子形成細胞因子風暴,短時間導致多器官功能衰竭,從而危及生命。HLH的病因及發病機制十分復雜,遺傳因素被認為是其發病的主要原因之一。依照HLH國際診療指南,通過基因檢測確認攜帶HLH易感基因突變的患者即可診斷為原發性HLH,造血干細胞移植是其唯一的治愈手段。目前用于臨床篩查的HLH易感基因有12個(PRF1等)。然而,約80%的病人檢測不到這些HLH基因突變,其中卻有相當一部分呈現家族聚集性、病情反復等原發HLH的典型特征。這些患者很可能存在尚未發現的遺傳學異常而延誤治療導致預后不良。因此,在已發現的12個HLH基因之外,是否還存在未知的致病遺傳突變?這些突變導致HLH的分子發病機制又是什么?
中國科學院北京基因組研究所(國家生物信息中心)王前飛、劉欣團隊與首都醫科大學附屬北京兒童醫院張蕊團隊合作,聚焦上述臨床問題,開展HLH新致病遺傳變異研究,結合家系模型、基因組突變、病人臨床表征及分子生物學功能驗證,突破性地發現了有助于臨床實際應用的HLH新致病基因。研究成果以“NBAS, a gene involved in cytotoxic degranulation, is recurrently mutated in pediatric hemophagocytic lymphohistiocytosis”為題,于2022年7月28日發表在Journal of Hematology & Oncology期刊。
研究團隊以未攜帶已知致病突變的高度疑診原發HLH病人為研究對象,對13例患兒及其父母進行全基因組或全外顯子組測序,通過生物信息學分析將潛在致病因素鎖定至病人中重復出現突變的NBAS基因。研究人員進一步在中國HLH隊列(224例散發HLH患兒)中驗證了NBAS基因突變的重現性,發現其突變發生率(2.11%)僅低于最常見的HLH突變基因PRF1(3.80%)。通過病人原代細胞表型以及體外細胞系功能實驗,證實了NBAS基因缺陷可能通過引起NK等免疫細胞的細胞毒功能失調產生HLH。
該研究基于大樣本中國HLH患病人群,結合家系模型對先天遺傳缺陷明顯的HLH患兒進行深入研究,在國際上首次發現HLH的全新致病基因NBAS,并結合功能實驗揭示了該基因在HLH中的內在分子機制。這項研究成果拓展了對HLH發病機制的理解,NBAS有望成為第13個HLH臨床診斷基因,最終將有助于臨床篩查HLH高危人群,提高病人的早診早治率。
中國科學院北京基因組研究所(國家生物信息中心)王前飛研究員、劉欣研究員以及首都醫科大學附屬北京兒童醫院血液科張蕊主任為本文共同通訊作者,課題組博士畢業生畢小慢、陳蕾和首都醫科大學附屬北京兒童醫院副研究員張晴為本文的共同第一作者。該研究得到了國家重點研發計劃、國家自然科學基金、北京市自然科學基金等項目資助。
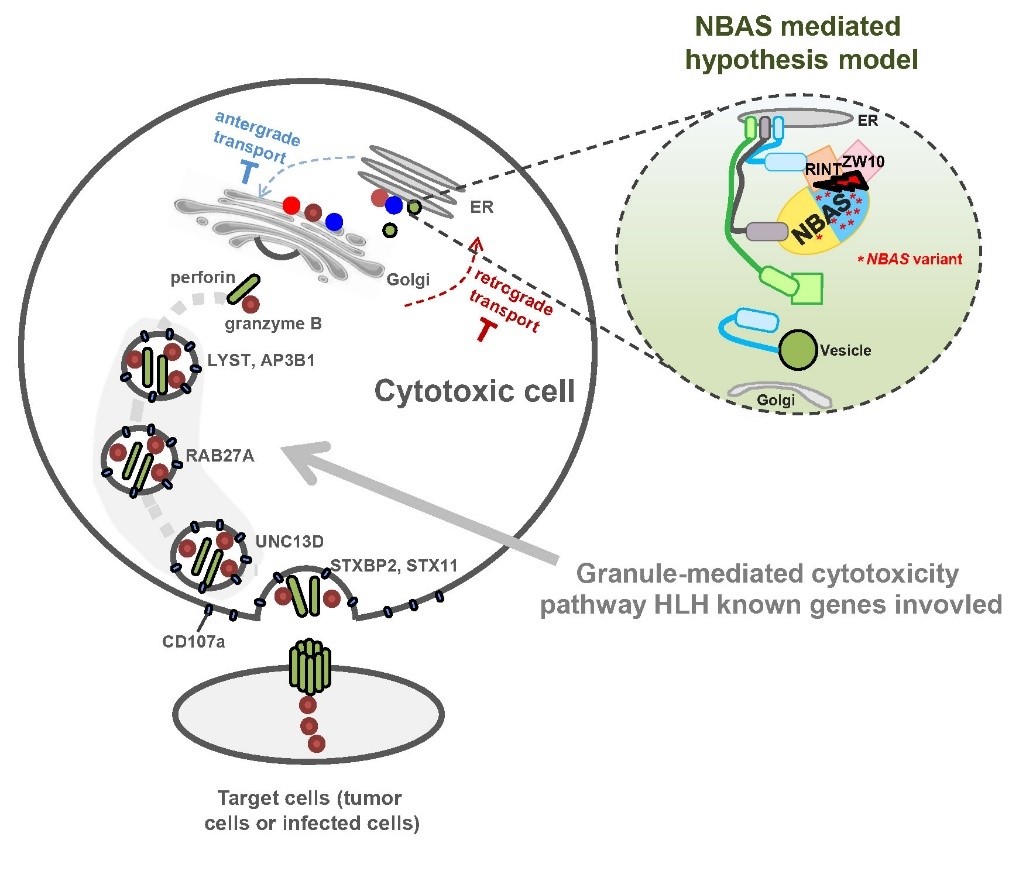
NBAS基因缺陷引起HLH發生的假說模型


